Dr Ali Alnasif recently completed his PhD in Mechanical Engineering at Cardiff University, where his research focused on developing an optimised kinetic reaction mechanism for ammonia–hydrogen combustion. His work examined why existing mechanisms often disagree in predicting flame speed, nitric oxide (NO) and nitrous oxide (N₂O) formation, and other key combustion features, and how these differences influence modelling accuracy under realistic, turbulence-affected flow conditions.
Dr Ali Alnasif recently completed his PhD in Mechanical Engineering at Cardiff University, where his research focused on developing an optimised kinetic reaction mechanism for ammonia–hydrogen combustion. His work examined why existing mechanisms often disagree in predicting flame speed, nitric oxide (NO) and nitrous oxide (N₂O) formation, and other key combustion features, and how these differences influence modelling accuracy under realistic, turbulence-affected flow conditions.
By analysing ammonia decomposition pathways, radical interactions and the sensitivity of NO-forming routes, he developed a compact and computationally efficient ammonia/hydrogen (NH₃/H₂) mechanism that delivers reliable chemical predictions while significantly reducing simulation time.
In this blog, he outlines the scientific motivation, methodology and practical impact of this mechanism-optimisation study, highlighting how improved chemical models can support cleaner and more accurate ammonia-based combustion technologies.
Why chemical reaction mechanisms matter in ammonia combustion
In combustion research, a reaction mechanism acts like a detailed map showing how fuel molecules break apart and change during burning. This “map” is important because it guides scientists in understanding how a fuel behaves and how accurately computer models can match experiments.
For ammonia-based fuels, a good reaction mechanism needs to describe the key steps in how ammonia decomposes, how reactive radicals are formed, and how nitrogen-containing compounds evolve during burning. It should also be able to reproduce important measurements, such as how quickly the fuel ignites, how fast a flame spreads, and which chemical species appear in the process.
How ammonia breaks down during combustion
When ammonia burns, it doesn’t transform into its final products in a single step. Instead, it moves through a series of small chemical reactions, described by the reaction mechanism, and forms short-lived intermediate molecules along the way. Some of these reaction routes lead to the formation of nitrogen oxides (NOₓ), which are pollutants, while others convert the fuel into harmless nitrogen gas (N₂).
The diagram below summarises these pathways.
- The red arrows show the routes that tend to produce NOₓ.
- The blue arrows highlight the routes that reduce NOₓ and help form N₂.
You don’t need to follow every detail in the figure. The key idea is that ammonia can behave in two different ways during combustion: it can either produce unwanted emissions or follow cleaner routes. The initial conditions, such as pressure, temperature, air-to-fuel ratio and the mixing proportion of NH₃ and H₂ in the fuel; play a major role in determining which pathway becomes dominant. Understanding these influences helps scientists design burners that are more efficient and generate fewer pollutants.
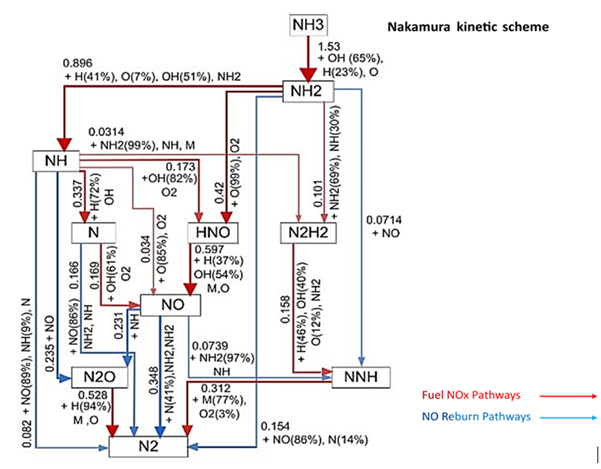
According to Alnasif et al. (2023), the diagram above shows how ammonia breaks down in a flame and highlights the main chemical steps that either produce nitrogen oxides (NOₓ) or help remove them. The arrows represent different reaction routes, and their thickness indicates how strongly each route contributes. The percentages show how much each chemical step influences the next stage in the process. These pathways are predicted using the Nakamura reaction mechanism under typical combustion conditions.
Why reaction mechanisms matter in real combustion simulations
Scientists gain the most value from reaction mechanisms when these mechanisms are combined with realistic flow-simulation tools. In practical studies of how combustion behaves inside real systems, researchers often use computational fluid dynamics (CFD). In this environment, the reliability of the reaction mechanism becomes even more important.
Turbulence, the chaotic motion of gases inside a flame, strongly influences how the fuel and air mix, how heat is distributed, how chemicals are transported, and how fast reactions occur. These effects create complex interactions between the flow field and the chemistry. Because of this, only reaction mechanisms with strong and well-validated chemical foundations can give trustworthy predictions when they are used together with realistic CFD models.
What makes a reaction mechanism reliable for ammonia combustion
For a reaction mechanism to be useful in real combustion studies, it needs to include a strong and dependable description of how hydrogen and oxygen react. This part of the chemistry controls the creation of highly reactive radicals that keep the flame going. The mechanism must also describe, with good accuracy, how ammonia and hydrogen interact as they burn.
The way hydrogen- and oxygen-based radicals interact with nitrogen-containing radicals, such as NHₓ, NOₓ, and N₂O, is particularly important. These interactions shape how the mixture of radicals develops inside the flame and directly influence which nitrogen species are formed, including pollutants.
At the same time, the mechanism must be efficient enough for use in detailed CFD simulations. If it is too large or too slow to compute, it becomes impractical for high-fidelity modelling of real combustion systems.
Why not all reaction mechanisms agree: the case of NO formation
Different reaction mechanisms can produce different predictions in combustion simulations because each mechanism describes the underlying chemistry using its own set of reactions, assumptions, and reaction rates. In other words, even when the same fuel and operating conditions are used, the predicted flame behaviour can vary depending on how the mechanism represents the chemistry inside the flame.
A clear example of this is the prediction of nitrogen oxide (NO). NO formation depends on a chain of fast and highly sensitive chemical steps involving ammonia breakdown, radical production, and nitrogen–oxygen interactions. If a mechanism includes more detailed pathways for forming NO or uses faster reaction rates for those steps, it will naturally predict higher NO levels. If another mechanism places more emphasis on reactions that convert NO back into harmless nitrogen gas (N₂), its predictions will be lower.
These differences occur because each mechanism highlights different parts of the chemistry. Some mechanisms include extensive nitrogen sub-mechanisms, detailed radical interactions, and multiple NO-reduction pathways, while others simplify these routes or use older reaction data. As a result, the chemical “image” of the flame is not identical from one mechanism to another, and this leads to noticeable variations in their predictions.
Understanding how these chemical choices shape the results helps researchers decide which mechanism is more reliable for modelling ammonia–hydrogen combustion in practical systems.
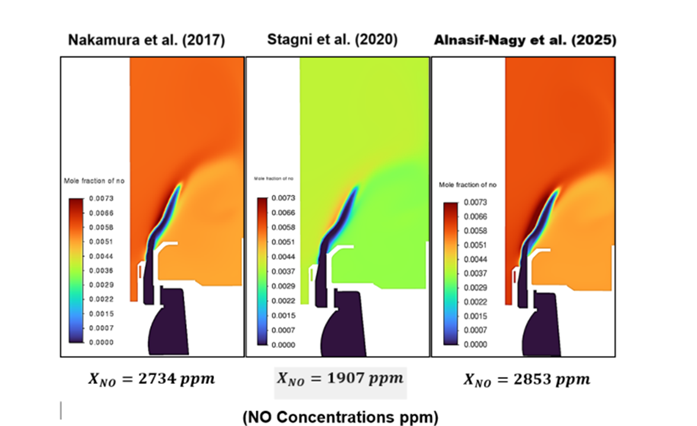
Contour plots showing the predicted NO concentrations in a swirl burner operating with a 70/30 vol% NH₃/H₂ fuel blend at an equivalence ratio of φ = 0.8. The images compare results from CFD simulations using three different reaction mechanisms: Nakamura et al. [2017], Stagni et al. [2020], and Alnasif -Nagy Mechanism. The variation in colour indicates how NO levels change across the flame region, highlighting how each mechanism produces different emission predictions under the same operating conditions.
Why computational efficiency matters in combustion modelling
Computational efficiency is also essential to ensure that a reaction mechanism remains practical for use in high-fidelity CFD studies. Full, highly detailed mechanisms often become too slow for real engineering simulations, especially in multidimensional CFD, where thousands of chemical reactions must be solved at every point in the flow. To overcome this challenge, reduced and optimised mechanisms are designed to keep strong predictive accuracy while greatly lowering the computational cost.
The compact NH₃/H₂ mechanism developed by Alnasif et al (2025) achieves reliable predictions of NO and N₂O by refining both the H₂/O₂ and NHₓ sub-mechanisms, allowing the chemistry to be solved more quickly and efficiently. According to the published work by Alnasif et al. (2025), the improved Alnasif–Nagy mechanism was tested against two well-known models: Nakamura et al. (2017) and Stagni et al. (2020). The study reported that the Alnasif–Nagy mechanism delivered a 2.14× speed-up compared with the Stagni mechanism and a 1.78× speed-up relative to the Nakamura model.
In practical terms, the total simulation time for a single CFD case, using 96 CPU cores, was 7.5 hours with the Alnasif–Nagy mechanism, compared with 16 hours for the Stagni mechanism and 13 hours for the Nakamura mechanism. This improvement in computational performance is especially valuable for reducing the time and resources required for CFD simulations.
References
[1] Alnasif, A., Mashruk, S., Hayashi, M., Jójka, J., Shi, H., Hayakawa, A., & Valera-Medina, A. (2023). Performance Investigation of Currently Available Reaction Mechanisms in the Estimation of NO Measurements: A Comparative Study. Energies, 16(9), 3847
[2] H. Nakamura, S. Hasegawa, T. Tezuka, Kinetic modeling of ammonia/air weak flames in a micro flow reactor with a controlled temperature profile, Combust. Flame 185 (2017) 16–27. https://doi.org/10.1016/j.combustflame.2017.06.021
[3] A. Stagni, C. Cavallotti, S. Arunthanayothin, Y. Song, O. Herbinet, F. Battin-Leclerc, T. Faravelli, An experimental, theoretical and kinetic-modeling study of the gas-phase oxidation of ammonia, React. Chem. Eng. 5 (2020) 696–711. https://doi.org/10.1039/c9re00429g
[4] Alnasif, A., Jojka, J., Papp, M., Szanthoffer, A. G., Kovaleva, M., Turányi, T., Mashruk, S., Valera-Medina, A., & Nagy, T. (2025). A compact kinetic reaction mechanism for NH3/H2 flames. Journal of Ammonia Energy, 3(1).